










苏州三点净化 医疗器械 GMP 车间解决方案
1、在生产或使用中活性物质、灭活物质的污染(包括热原)对产品产生重要影响的植入性医疗器械,医疗器械 GMP 车间规划设计,应对工作环境进行控制,对灭活的方法应予验证并保存记录。此类产品的生产和包装应在有规范要求的、可控的环境下进行。
2、对非无菌植入性医疗器械或使用前预期灭菌的医疗器械,如果通过确认的产品清洁、包装过程,能将污染降低并保持一致的控制水平,医疗器械 GMP 车间施工,应建立一个受控的环境来包含该确认的清洁和包装过程。生产企业可参照 YY0033-2000 标准或自行验证并确定产品的生产洁净级别。
3、应对受污染或易于污染的产品进行控制。应对受污染或易于污染的产品、工作台面或人员建立搬运、清洁和除污染的文件。医疗器械 GMP 车间施工设计参考
1、《医疗器械生产企业质量管理规范(试行)》,国家食品药品监督管理局(2009 年)--2015 废止。
2、《体外诊断试剂生产实施细则(试行)》,国家食品药品监督管理局(2007 年)--2015 废止。
3、《关于实施 ( 医疗器械生产质量管理规范(试行)) 及其配套文件有关问题的通知》(2011 年)--2015 废止。来东净化补充说明前面 1~3 的 2007、2009 年医疗器械的规范、细则、标准在 2015年停用,医疗器械 GMP 车间规划,代之 2015 的医疗器械生产企业质量管理规范及无菌、植入、体外诊断试剂三个附录。
5、《无菌医疗器具生产管理规范》(YY0033-2000)
6、《洁净厂房设计规范》(GB50073-2010)
7、《洁净室施工及验收规范》(GB50591-2010)……
19、《医 疗 产 品 的 无 菌 加 工 第 1 部 分:通 用 要 求》(YY/T0567.1-2005)
20、《无菌医疗器械生产与质量管理讲义》,国家药品监督管理局(2000)
23、《无菌医疗器械质量控制与评价》,苏州大学出版社(2012 年)
24、《无菌医疗器械生产与洁净厂房的建设》,CMD(2009 年)
医疗器械 GMP 车间工程设计的规范参照:
1、国际标准《ISO/DIS14644》
2、洁净室厂房设计规范《GB50073-2001》
3、医疗器械包装车间洁净室厂房规范《GMP-97》
4、药品生产质量管理规范《GMP-98》
5、洁净室施工及难收规范《JGJ71-90》
6、通风与空调工程施工及验收规范《GB50243-2002》
7、美国联邦标准《FS209E-92》
医疗器械净化工程建设中需考虑从以下问题:
1. 医疗器械包装车间洁净室工程所需要的净化材料;
2. 医疗器械厂房洁净室及医疗器械包装车间洁净室工程的设计、安装、调试、维护等综合服务;
3. 医疗器械包装车间洁净室工程空调净化部分。温度和相对湿度无菌医疗器械在无特殊规定时,通常要求温度在法规标准检测StandardandTesting18~28C,湿度在 45%~65%,企业一般都可以控制在要求内。如在动态监测中发现达不到要求,可能是室内有产热大的仪器设备。风量、换气次数、静压差在洁净室体积确定的情况下,换气次数由该室的送风量决定,而静压差取决于房间的送风量与回风量、排风量的差值。系统总送风量、新风量、总排风量和对外压差可以通过调整风机频率转速或总阀门开启度来实现,各房间的风量和压力则可通过调整分支管路阀门开度来实现。悬浮粒子、浮游菌、沉降菌测试条件如不能满足规定的环境参数 ( 温湿度、风速、换气次数、静压差在规定范围之内 ) 要求,关键项目悬浮粒子、浮游菌或沉降菌的测试结果应视为无效。由于温度、相对湿度、风速、换气次数、静压差共同构成了洁净室的微气候,是洁净室维护正常与否的重要指征,可将关键工序关键项目测试修订为关键工序全性能测试。
温度
洁净室夏季室温超过设计范围的原因,多是由于开始确定的各洁净室的空调送风量即换气次数时只注重满足洁净度指标,忽视了对各洁净室热平衡的校核计算。因此在生产洁净室的设计及运行过程中,必须对洁净室的空调送风参数进行实时修正,保证各个季节生产洁净室的温度都维持 18~28C。温度和相对湿度主要影响产品生产工艺及细菌的繁殖条件,还能引发由生产操作人员舒适度对产品质量的影响。送风量、换气次数医疗器械净化工程 - 无菌洁净室工程设计阶段对送风量的确定,首先要满足相应洁净度级别的换气次数要求,同时还要通过热、湿负荷校核来进一步确定风量,在此基础上对高效过滤器进行选用。过滤器的处理风量应小于或等于额定风量,设置在同一洁净区内的高效 ( 亚高效、超高效 ) 空气过滤器的阻力、效率宜接近。
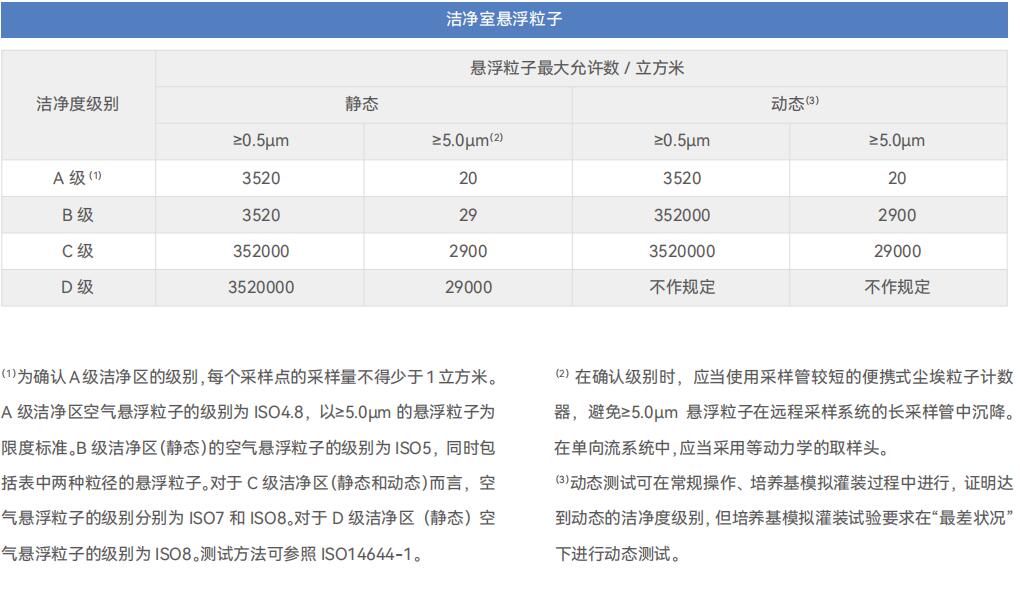 医疗器械 GMP 车间管控总要求:
医疗器械 GMP 车间管控总要求:
(1)表面平滑;
(2)表面有耐磨性;
(3)良好的热绝缘性;
(4)不易产生静电;
(5)不吸湿,不透湿;
(6)吸声性好;
(7)容易加工;
(8)表面不易附着灰尘;
(9)容易除去附着的灰尘;
1、在生产或使用中活性物质、灭活物质的污染(包括热原)对产品产生重要影响的植入性医疗器械,医疗器械 GMP 车间规划设计,应对工作环境进行控制,对灭活的方法应予验证并保存记录。此类产品的生产和包装应在有规范要求的、可控的环境下进行。
2、对非无菌植入性医疗器械或使用前预期灭菌的医疗器械,如果通过确认的产品清洁、包装过程,能将污染降低并保持一致的控制水平,医疗器械 GMP 车间施工,应建立一个受控的环境来包含该确认的清洁和包装过程。生产企业可参照 YY0033-2000 标准或自行验证并确定产品的生产洁净级别。
3、应对受污染或易于污染的产品进行控制。应对受污染或易于污染的产品、工作台面或人员建立搬运、清洁和除污染的文件。医疗器械 GMP 车间施工设计参考
1、《医疗器械生产企业质量管理规范(试行)》,国家食品药品监督管理局(2009 年)--2015 废止。
2、《体外诊断试剂生产实施细则(试行)》,国家食品药品监督管理局(2007 年)--2015 废止。
3、《关于实施 ( 医疗器械生产质量管理规范(试行)) 及其配套文件有关问题的通知》(2011 年)--2015 废止。来东净化补充说明前面 1~3 的 2007、2009 年医疗器械的规范、细则、标准在 2015年停用,医疗器械 GMP 车间规划,代之 2015 的医疗器械生产企业质量管理规范及无菌、植入、体外诊断试剂三个附录。
5、《无菌医疗器具生产管理规范》(YY0033-2000)
6、《洁净厂房设计规范》(GB50073-2010)
7、《洁净室施工及验收规范》(GB50591-2010)……
19、《医 疗 产 品 的 无 菌 加 工 第 1 部 分:通 用 要 求》(YY/T0567.1-2005)
20、《无菌医疗器械生产与质量管理讲义》,国家药品监督管理局(2000)
23、《无菌医疗器械质量控制与评价》,苏州大学出版社(2012 年)
24、《无菌医疗器械生产与洁净厂房的建设》,CMD(2009 年)
医疗器械 GMP 车间工程设计的规范参照:
1、国际标准《ISO/DIS14644》
2、洁净室厂房设计规范《GB50073-2001》
3、医疗器械包装车间洁净室厂房规范《GMP-97》
4、药品生产质量管理规范《GMP-98》
5、洁净室施工及难收规范《JGJ71-90》
6、通风与空调工程施工及验收规范《GB50243-2002》
7、美国联邦标准《FS209E-92》
医疗器械净化工程建设中需考虑从以下问题:
1. 医疗器械包装车间洁净室工程所需要的净化材料;
2. 医疗器械厂房洁净室及医疗器械包装车间洁净室工程的设计、安装、调试、维护等综合服务;
3. 医疗器械包装车间洁净室工程空调净化部分。温度和相对湿度无菌医疗器械在无特殊规定时,通常要求温度在法规标准检测StandardandTesting18~28C,湿度在 45%~65%,企业一般都可以控制在要求内。如在动态监测中发现达不到要求,可能是室内有产热大的仪器设备。风量、换气次数、静压差在洁净室体积确定的情况下,换气次数由该室的送风量决定,而静压差取决于房间的送风量与回风量、排风量的差值。系统总送风量、新风量、总排风量和对外压差可以通过调整风机频率转速或总阀门开启度来实现,各房间的风量和压力则可通过调整分支管路阀门开度来实现。悬浮粒子、浮游菌、沉降菌测试条件如不能满足规定的环境参数 ( 温湿度、风速、换气次数、静压差在规定范围之内 ) 要求,关键项目悬浮粒子、浮游菌或沉降菌的测试结果应视为无效。由于温度、相对湿度、风速、换气次数、静压差共同构成了洁净室的微气候,是洁净室维护正常与否的重要指征,可将关键工序关键项目测试修订为关键工序全性能测试。
温度
洁净室夏季室温超过设计范围的原因,多是由于开始确定的各洁净室的空调送风量即换气次数时只注重满足洁净度指标,忽视了对各洁净室热平衡的校核计算。因此在生产洁净室的设计及运行过程中,必须对洁净室的空调送风参数进行实时修正,保证各个季节生产洁净室的温度都维持 18~28C。温度和相对湿度主要影响产品生产工艺及细菌的繁殖条件,还能引发由生产操作人员舒适度对产品质量的影响。送风量、换气次数医疗器械净化工程 - 无菌洁净室工程设计阶段对送风量的确定,首先要满足相应洁净度级别的换气次数要求,同时还要通过热、湿负荷校核来进一步确定风量,在此基础上对高效过滤器进行选用。过滤器的处理风量应小于或等于额定风量,设置在同一洁净区内的高效 ( 亚高效、超高效 ) 空气过滤器的阻力、效率宜接近。
(1)表面平滑;
(2)表面有耐磨性;
(3)良好的热绝缘性;
(4)不易产生静电;
(5)不吸湿,不透湿;
(6)吸声性好;
(7)容易加工;
(8)表面不易附着灰尘;
(9)容易除去附着的灰尘;

